- Teste de hematologie
- Teste de biochimie
- Biochimie generală din sânge și urina
- Proteine specifice in ser si urina
- Teste biochimice din lichide de punctie
- Teste biochimice din materii fecale
- Teste biochimice pentru tulburari ereditare de metabolism
- Teste pentru nefrolitiaza
- Vitamine, oligoelemente, stres oxidativ
- Acizi grași
- Transferina carbohidrat deficitara (CDT) marker pentru alcoolism
- Markeri non-invazivi pentru afecţiunile hepatice
- Analiza chimică calculi
- Markeri endocrini
- Markeri tumorali
- Markeri virali
- Markeri cardiaci
- Markeri anemie
- Markeri ososi
- Markeri boli autoimune
- Anticorpi antispermatozoizi
- Autoanticorpi in afectiuni endocrine, cardiace, renale
- Autoanticorpi in afectiuni neurologice
- Autoanticorpi in afectiunile dermatologice
- Autoanticorpi in anemia pernicioasa
- Autoanticorpi in diabetul zaharat
- Markeri pentru afectiuni hepatice si gastrointestinale autoimune
- Markeri pentru afectiuni reumatismale si vasculite
- Markeri pentru monitorizarea evolutiei si tratamentului
- Markeri pentru sindromul antifosfolipidic
- Serologie boli infectioase
- Teste specializate de alergologie si imunologie
- Teste de biologie moleculara
- Teste de citogenetica
- Teste de microbiologie
- Toxicologie
- Citologie cervico-vaginala
- Histopatologie
- Uncategorized

Screening metabolic neonatal extins
Informații generale și recomandări
Screening-ul neonatal, o componentă importantă a medicinei preventive, include un panel de teste pentru identificarea precoce a unor condiții patologice la care intervenția în timp util are drept consecință eliminarea sau reducerea morbidității, mortalității și a dizabilităților.
De la prima inițiativă de screening neonatal din anii 1960 pentru depistarea fenilcetonuriei numărul afecțiunilor pentru care s-au dezvoltat teste de screening în perioada neonatală a crescut dramatic, în special în urma introducerii analizei multiplex prin spectrometrie de masă în tandem a profilelor de aminoacizi și acilcarnitină1.
Tulburările ereditare de metabolism au suscitat atenția neonatologilor și pediatrilor prin faptul că multe din aceste afecțiuni pot fi tratate eficient dacă sunt detectate precoce si de aici importanța includerii acestora în testele de screening din perioada neonatală. Pe altă parte, chiar dacă unele boli nu beneficiază de tratament screening-ul lor este de asemenea justificat, pentru a facilita diagnosticul prenatal la sarcinile viitoare2.
Tulburările ereditare de metabolism reprezintă un grup de afecțiuni monogenice ce afectează funcția unor proteine cu rol esențial în diverse căi metabolice (enzime, cofactori, receptori, transportori, etc) . Anomaliile biochimice rezultate conduc la fie la acumularea unor metaboliți intermediari sau de by-pass, fie la o deficiență de metaboliți terminali generând tulburări metabolice severe, cum ar fi acidoza lactică, cetoacidoza sau hiperamoniemia3. Anomaliile biochimice pot afecta aminoacizii, acizii organici, carbohidrații, acizii grași, purinele și pirimidinele. Până în prezent au fost identificate peste 500 de afecțiuni metabolice ereditare, fiind estimată o incidență cumulată globală ce variază de la 1/800 la 1/2500 nou-născuți4.
Majoritatea metaboliților anormali asociați cu tulburările ereditare de metabolism sunt excretați în urină. Având în vedere faptul că probele de urină sunt ușor de colectat pe hârtie de filtru sunt preferate pentru testele de screening neonatal5.
Metabolomica reprezintă o nouă abordare pentru analiza modificărilor suferite de toți compușii cu greutate moleculară intermediară și metaboliții terminali. Gaz-cromatografia cuplată cu spectrometria de masă (GC/MS) constituie un instrument analitic avantajos pentru studiile de metabolomică fiind capabilă să separe și să cuantifice simultan câteva sute de metaboliți, într-un timp scurt. Ca principiu general, extractele de probe biologice sunt supuse volatilizării înainte de separarea printr-o coloană capilară, cu o fază staționară adecvată pentru analiza compușilor de interes. Compușii separați sunt eluați într-un spectrometru de masă echipat cu o sursă de ionizare prin impact electronic, fiind generate spectre de masă cuantificabile6.
În 1966, Tanaka et al de la Massachussets General Hospital au detectat pentru prima oară acidemia izovalerică prin GC/MS evidențiind importanța acestei tehnici. În 1977 a fost inițiat în Japonia un program de depistare a 22 afecțiuni metabolice. Mai târziu, în 1991, Shoemaker și Eliott de la Universitatea St. Louis, USA au raportat că acizii organici, aminoacizii și carbohidrații pot fi separați eficient în urină prin GC/MS. Procedura analitică aplicată de aceștia era însă laborioasă astfel că, în 1996 a fost simplificată de Kuhara et al. În 2007 tehnica GC/MS a fost utilizată pentru detectarea fenilalaninei, tirozinei, leucinei și valinei din spoturi de sânge la nou-născuți. Screening-ul neonatal utilizand probe de sânge recoltate pe carduri Guthrie a fost introdus cu succes în SUA și țările dezvoltate din Europa. Ulterior, alături de sânge au început să fie folosite și probe de urină. Deoarece compușii toxici pentru organism sau cei care depășesc nivelul optim în sânge sunt excretați prin urină în cantități mari pot fi detectați cu ușurință prin GC/MS. În plus, colectarea probelor de urină este neinvazivă comparativ cu recoltarea probelor de sânge. Progresul înregistrat recent în metodologia de separare în asociere cu spectrometria de masă a crescut eficiența GC/MS și a îmbunătățit remarcabil detectarea tulburărilor ereditare de metabolism prin screening neonatal7.
Testul Baby Sensor 100+ (Novogenia, Austria) disponibil în laboratoarele Synevo oferă un screening neonatal pentru 100 afecțiuni metabolice ereditare (aminoacidopatii, acidemii organice, disfuncții ale ciclului Krebs/anomalii mitocondriale, boli peroxizomale, tulburări în metabolismul carbohidraților, acizilor grași, purinelor și pirimidinelor) și, în plus, față de neuroblastom. Este raportată o rată de detecție de 99%. Sunt necesare probe de urină spontană recoltate pe hârtie de filtru.
În tabelul de mai jos sunt prezentate afecțiunile ce pot fi detectate cu acest test, complicațiile acestora și tipul de intervenție disponibil8:
| Denumire afecțiune | Complicații | Tip de intervenție |
| Galactozemia tranzitorie | Retard de creștere și disfuncție hepatică | Tip A |
| Aciduria 2-cetoadipică | Retard de dezvoltare și alte probleme neurologice | Tip A |
| Aciduria 3-hidroxi, 3-metil glutarică (deficitul de 3-hidroxi 3-metilglutaril CoA liază) | Acidoză metabolică severă fără cetoză și retard de dezvoltare | Tip A |
| Deficiența de 3-hidroxi-izobutiril CoA decarboxilază | Hipoglicemie hipoketotică severă și afectare neurologică | Tip B |
| Deficiența de 3-metilcrotonil CoA carboxilază | Acidoză metabolică acută, retard de dezvoltare | Tip A |
| Aciduria 3-metil glutaconică | Cardiomiopatie și miopatie ce afectează musculatura scheletică | Tip B |
| 5-oxoprolinuria | Convulsii, retard mental, lipsa coordonării | Tip A |
| Hiperglicinuria (cetotică) | Retard de creștere | Tip A |
| Deficiența de adenin-fosforibozil transferază | Infecții de tract urinar | Tip B |
| Deficiența de adenozin deaminază | Otită și infecții de tract respirator recurente | Tip B |
| Alcaptonuria | Afectarea cartilajelor | Tip A |
| Argininemia | Retard de dezvoltare | Tip A |
| Aciduria arginino-succinică | Retard mental și motor | Tip A |
| Deficiența de biotinidază | Rash facial/perioral, retard mental | Tip A |
| Boala Canavan | Retard sever de dezvoltare | Tip B |
| Acidemia hiperurică | Surditate neurosenzorială, urolitiază | Tip A |
| Citrulinemia | Letargie și comportament anormal | Tip A |
| Cistationuria | Disfuncție hepatică | Tip A |
| Cistinurie | Retard de dezvoltare | Tip A |
| Hiperfenilalaninemia benignă | Eczemă, păr și piele de culoare deschisă | Tip A |
| Aciduria metilmalonică, formele cblA, cblB | Hipotonie, retard de dezvoltare, letargie, hepatomegalie | Tip A |
| Aciduria D-glicerică | Retard de creștere | Tip C |
| Sucrozuria endogenă | Retard mental | Tip B |
| Deficiența de dihidropiridinază | Convulsii neonatale | Tip D |
| Deficiența de dihidrolipoil dehidrogenază (E3) | Miros dulceag al urinei și corpului (de zahăr ars) | Tip D |
| Aciduria etilmalonică | Retard de creștere /comă | Tip D |
| Aciduria formiminoglutamică | Anemia megaloblastică | Tip A |
| Deficiența de fructozo-1,6-difosfatază | Hipoglicemie cu cetoză | Tip A |
| Deficiența de fumarat hidratază | Convulsii cu retard sever | Tip D |
| Galactozemia | Disfuncție hepatică | Tip A |
| Aciduria glutarică tip I | Macrocefalie cu dificultăți de mișcare | Tip A |
| Aciduria glutarică tip II | Miros de picioare transpirate, tulburări respiratorii, retard mental și motor | Tip B |
| Glutationuria | Anemie hemolitică | Tip B |
| Deficiența de beta-ketotiolază | Vărsături, deshidratare, dificultăți de respirație, letargie și convulsii (ocazional) | Tip A |
| Hiperoxalurie primară tip 2 | Litiază renală, insuficiență renală, afectarea altor organe | Tip A |
| Boala Hartnup | Fotosensibilitate, anomalii oculare | Tip A |
| Hawkinsonuria | Disfuncție hepatică | Tip C |
| Histidinemia | Retard mental, anomalii renale | Tip C |
| Histidinuria | Retard mental, dismorfism facial | Tip A |
| Homocistinuria | Retard mental | Tip A |
| Hidroxilizinuria | Retard mental, tulburări de comportament, hiperactivitate | Tip C |
| Hiperhidroxiprolinemia | Retard mental | Tip C |
| Hiperglicinuria | Retard de creștere | Tip B |
| Hiperleucin-izoleucinemia | Convulsii, retard de creștere | Tip B |
| Hipermetioninemia | Facies neobișnuit, tulburări neurologice, retard motor | Tip A |
| Sindromul de hiperornitinemie, hiperamoniemie, hiperhomocitrulinemie (HHH) | Vărsături, letargie, retard de dezvoltare, tulburări de învățare | Tip D |
| Hiperpipecolatemia | Retard sever de dezvoltare | Tip C |
| Hiperprolinemia tip I | Tulburări neurologice sau psihiatrice | Tip C |
| Hiperprolinemia tip II | Convulsii, retard mental | Tip C |
| Aminoaciduria imidiazolica | Convulsii, retard de dezvoltare | Tip B |
| Iminoglicinuria | Retard mental, manifestări renale | Tip C |
| Boala Refsum infantilă | Cecitate și tulburări de auz/retinită pigmentară | Tip C |
| Acidemia izovalerică | Spasme datorate hipotermiei, miros de picioare transpirate | Tip A |
| Intoleranța la lactoză | Retard de dezvoltare | Tip A |
| Sindromul Lesch-Nyhan | Retard mental, automutilări | Tip C |
| Deficiența de „long-chain” acil CoA dehidrogenază (LCAD) | Slăbiciune musculară, mialgii persistente | Tip A |
| Lizinuria | Retard mental | Tip C |
| Intoleranța la proteina lizinurică | Retard de creștere | Tip B |
| Boala urinei cu miros de arțar | Tulburări neurologice, letargie miros caracteristic al urinei (zahăr ars) | Tip A |
| Deficiența de „medium-chain” acil CoA dehidrogenază (MCAD) | Retard de creștere | Tip A |
| Acidemia metilmalonică (MMA) | Convulsii, letargie | Tip A |
| Acidemia metilmalonică datorată metabolismului anormal, absorbției și transportului vitaminei B12 | Slăbiciune musculară, convulsii, anemie | Tip A |
| Deficiența multiplă de carboxilază (MCD) | Acidoză metabolică, hipotonie, retard de dezvoltare | Tip A |
| Acidemia mevalonică | Dismorfism cranian, retard de dezvoltare | Tip B |
| Deficiența de metilmalonic semialdehid dehidrogenază (MMSDH) | Acidoză metabolică,, letargie, convulsii | Tip B |
| Deficența de N-acetilglutamat sintetază (NAGS) | Complicații neurologice | Tip B |
| Adrenoleucodistrofia neonatală | Slăbiciune musculară | Tip D |
| Neuroblastom | Regresie spontană | Tip A |
| Colestaza intrahepatică neonatală cauzată de deficiența de citrină (NICCD) | Disfuncție hepatică | Tip A |
| Deficiența de ornitintranscarbamilază (OTC) | Comportament iritativ, retard de dezvoltare | Tip A |
| Aciduria orotică | Malformații cardiace, anemie | Tip B |
| Deficiența parțială de hipoxantin-adenin fosforibozil transferază | Litiază renală, tulburări de mișcare | Tip D |
| Fenilcetonuria (PKU) | Retard de dezvoltare, tulburări de comportament | Tip A |
| Hiperoxaluria primară | Colici renale asociate cu litiazei | Tip A |
| Acidemia propionică | Letargie, tulburări de nutriție, hipotonie | Tip A |
| Deficiența de glicerolkinază | Retard de creștere | Tip A |
| Deficiența de piruvat carboxilază | Tulburări respiratorii | Tip D |
| Deficiența de piruvat decarboxilază | Retard de creștere, retard psihomotor sever cu tulburări de vedere | Tip D |
| Deficiența de piruvat dehidrogenază (E1) | Tulburări de nutriție, letargie și tulburări respiratorii | Tip D |
| Deficiența de piruvat dehidrogenază fosfatază | Acidoză lactică cu hipotonie | Tip D |
| Zaharopinuria | Trăsături dismorfice, statură redusă | Tip C |
| Deficiența de carbamilfosfat sintetaza I | Complicații neurologice | Tip B |
| Tirozinemia tip III | Retard mental ușor, convusii, tulburări de echilibru | Tip B |
| Deficiența de carnozinază serică | Hipotonie, retard de dezvoltare | Tip C |
| Deficiența de „short-chain” acil CoA dehidrogenază (SCAD) | Hipoglicemie, letargie | Tip A |
| Citrulinemia tip II | Agitație, pierderea memoriei, comportament anormal, convulsii, comă | Tip B |
| Timin uraciluria | Retard mental | Tip D |
| Tirozinemia tranzitorie a nou-născutului | Icter prelungit, letargie | Tip A |
| Triptofanuria cu nanism | Nanism, retard mental | Tip B |
| Tirozinemia datorată disfuncției hepatice | Disfuncție hepatică | Tip A |
| Tirozinemia tip I | Miros carcateristic al urinei (de varză), disfuncție hepatică | Tip A |
| Tirozinemia tip II | Fotosensibilitate, retard de dezvoltare | Tip B |
| Valinemia | Retard de creștere, vărsături | Tip D |
| Xantinuria | Insuficiență renală acută | Tip A |
| Acidurie xanturenică | Retard mental | Tip A |
| Sindrom Zellweger-like | Hipotonie, retard psihomotor sever | Tip D |
| Sindrom Zellweger | Hipotonie, trăsături dismorfice | Tip D |
| Aciduria β-aminoizobutirică | Tulburări neurologice | Tip D |
| Defecte în biosinteza cofactorului biopterinei
(BIOPT BS) |
Retard de dezvoltare, epilepsie, tulburări de comportament | Tip B |
| Defecte în regenerarea cofactorului biopterinei
(BIOPT REG) |
Dizabilitate intelectuală, tulburări de mișcare, dificultăți de deglutiție, convulsii | Tip B |
| Deficiența de galactokinază (GALK) | Cataractă, hepatosplenomegalie | Tip B |
| Deficiența de galactozo-epimerază (GALE) | Retard de creștere și dezvoltare, dizabilitate intelectuală, afectare hepatică și renală | Tip B |
| Deficiența de izobutiril-CoA dehidrogenaza (IB) | Anemie, hipotonie, retard de dezvoltare | Tip B |
| Acidemia malonică (MAL) | Hipoglicemie, vărsături, diaree, convulsii | Tip B |
| Deficiența de metilmalonil-CoA mutază (MUT) | Cetoacidoză severă, disfuncție psihomotorie, distonie | Tip B |
| Deficiența proteinei trifuncționale mitocondriale | Letargie, hipoglicemie, hipotonie, afectare hepatică | Tip B |
| Fructozuria | Hepatomegalie, icter, ciroză, convulsii, retard de creștere și mental | Tip C |
| Hipersarcozinemia | Tulburări vizuale, cardiomiopatie, sinostoză craniană, retard de creștere și mental | Tip C |
| Deficiența de succinic semialdehid dehidrogenază | Hipotonie, hiporeflexie, convulsii, tulburări de mers neprogresive | Tip D |
| Histidinuria | Retard mental, defect de vorbire | Tip D |
| Deficiența de aminoacilază I | Surditate, slăbiciune musculară, atrofie cerebrală | Tip D |
Tipuri de intervenție:
Tip A – măsurile de prevenție și tratament sunt foarte eficiente pentru această afecțiune. În majoritatea cazurilor, copilul este sănătos sau prezintă complicații minore, dacă tratamentul se efectuează în timp util.
Tip B – Prevenția acestei afecțiuni este mai dificilă, însă diagnosticul corect este util pentru profilaxia și tratamentul complicațiilor acute cu potențial letal.
Tip C – În general, aceste afecțiuni sunt considerate benigne. Totuși, în aproximativ 10% din cazuri pot apărea complicații ce necesită intervenție medicală.
Tip D – Aceste afecțiuni sunt mai severe, cu impact semnificativ asupra sănătății copiilor, chiar dacă sunt diagnosticate precoce. Diagnosticul corect este important pentru condițiile de urgență în care este necesară o terapie de susținere pentru a reduce simptomele și complicațiile.
Recomandări pentru efectuarea screening-ului neonatal
- fiecărui copil, începând de la 48 ore după naștere, până la vârsta de 5 ani. După vârsta de 5 ani beneficiile pentru prevenția complicațiilor bolilor metabolice ereditare sunt limitate.
- suspiciune de afecțiune metabolică ereditară (screening selectiv): prezența de tulburări metabolice ca acidoza lactică, cetoacidoza, hiperamoniemia8.
Specimen recoltat – urină recoltată pe hârtie de filtru.
Kit-ul de recoltare este furnizat de către laborator. Se alege din kit-ul de recoltare pachetul cu codul de culoare roz. Celelalte seturi de prelevare a probelor nu sunt necesare.
În pachet se găsesc 3 hârtii de filtru. Se recomandă umezirea celor 3 hârtii de filtru cu urina copilului, dar în principiu, unul este suficient pentru analiză. Există 3 metode de recoltare a urinei care s-au dovedit a fi eficiente:
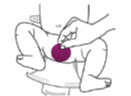 Recoltarea directă: se ține hârtia de filtru în fața zonei genitale a bebelușului până când aceasta este umezită cu urină. Această metodă necesită uneori puțină răbdare.
Recoltarea directă: se ține hârtia de filtru în fața zonei genitale a bebelușului până când aceasta este umezită cu urină. Această metodă necesită uneori puțină răbdare.
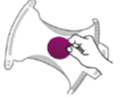
Metoda inserării în scutec: se plasează hârtia de filtru într-un scutec curat și se verifică la fiecare 30 minute. Dacă se găsește și scaun pe această hârtie de filtru, se aruncă hârtia și se încearcă din nou cu o altă bandă.

Metoda de înmuiere: se colectează 20-25 ml din urina copilului într-un recipient steril nou, curat (care nu a fost umplut, folosit sau clătit înainte) și se înmoaie hârtia de filtru în acesta.
1.Uscarea probei:
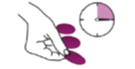
Se așază hârtia de filtru umezită pe o suprafață curată și se lăsă să se usuce la aer timp de 3 ore. A NU se utiliza dispozitive de uscare rapidă, precum uscătorul de par sau expunerea directă la soare pentru uscarea probei. Se pregătesc 3 eșantioane în acest fel dacă este posibil.
A NU se utiliza presarea hârtiei de filtru într-un scutec umed, deoarece urina este diluată si nu poate fi analizata în mod corespunzător. În acest caz nu se pot obține rezultate pentru testul Baby Sensor 100+.
2.Predarea probei: După uscarea completă a probei, aceasta se introduce în punga de plastic, se sigilează punga si se etichetează cu numele copilului.
Metodă – cromatografie de gaze cuplată cu spectrometrie de masă (GC/MS)8.
Raportarea și interpretarea rezultatelor – sunt analizați 250 metaboliți urinari iar profilul cromatografic (cromatograma) al probei este comparat cu profilul de referință (standard). Anumite combinații de metaboliți sunt sugestive pentru anumite afecțiuni metabolice. Sunt generate grafice cu valorile obținute la biomarkerii urinari raportate la valorile de referință precum și interpretări ale acestora pentru fiecare categorie de afecțiuni testate:
- Aminoacidopatii și acidemii organice;
- Disfuncții ale ciclului Krebs/anomalii mitocondriale
- Tulburări în metabolismul acizilor grași
- Boli peroxizomale
- Tulburări în metabolismul purinelor și pirimidinelor
- Tulburări în metabolismul carbohidraților
- Neuroblastom
Limite și interferențe
Administrarea unor medicamente sau a unor fluide în perfuzii poate determina creșteri false ale unor metaboliți urinari de aceea este esențial ca rezultatele obținute să fie corelate cu datele clinice.
Referințe
- Marzia Pasquali, Ph.D., F.A.C.M.G. and Nicola Longo, M.D., Ph.D. Newborn Screening and Inborn Errors of Metabolism. https://basicmedicalkey.com/newborn-screening-and-inborn-errors-of-metabolism, 2016. Reference Type: Internet Communication.
- Marsden D, Larson C, Levy HL. Newborn screening for metabolic disorders. J Pediatr. 2006 May;148(5):577-584.
- Pampols T. Inherited metabolic rare disease. Adv Exp Med Biol. 2010;686:397–431.
- Blau N, Duran M, Gibson KM, Dionisi-Vici C, editors. Physician’s Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases. Springer; Heidelberg: 2014.
- Janeckova H, Kalivodova A, Najdekr L, Friedecky D, Hron K, Bruheim P, Adam T. Untargeted metabolomic analysis of
- urine samples in the diagnosis of some inherited metabolic disorders. Biomed Pap Med Fac Univ Palacky Olomouc Czech
- Repub. 2015 Dec;159(4):582-5.
- De Souza DP. Detection of polar metabolites through the use of gas chromatography-mass spectrometry. Methods Mol
- Biol. 2013;1055:29-37. Sharma P, Kumar P, Sharma R, Dhot PS. Scope of gas chromatography-mass spectrometry in inborn errors of metabolism screening: A review. Asian Journal of Pharmaceutical and Clinical Research, Vol.8, Issue 1, 2015.
- Laborator Synevo. Referințele specifice tehnologiei de lucru utilizate 2017. Ref Type: Catalog.