- Teste de hematologie
- Teste de biochimie
- Biochimie generală din sânge și urina
- Proteine specifice in ser si urina
- Teste biochimice din lichide de punctie
- Teste biochimice din materii fecale
- Teste biochimice pentru tulburari ereditare de metabolism
- Teste pentru nefrolitiaza
- Vitamine, oligoelemente, stres oxidativ
- Acizi grași
- Transferina carbohidrat deficitara (CDT) marker pentru alcoolism
- Markeri non-invazivi pentru afecţiunile hepatice
- Analiza chimică calculi
- Markeri endocrini
- Markeri tumorali
- Markeri virali
- Markeri cardiaci
- Markeri anemie
- Markeri ososi
- Markeri boli autoimune
- Anticorpi antispermatozoizi
- Autoanticorpi in afectiuni endocrine, cardiace, renale
- Autoanticorpi in afectiuni neurologice
- Autoanticorpi in afectiunile dermatologice
- Autoanticorpi in anemia pernicioasa
- Autoanticorpi in diabetul zaharat
- Markeri pentru afectiuni hepatice si gastrointestinale autoimune
- Markeri pentru afectiuni reumatismale si vasculite
- Markeri pentru monitorizarea evolutiei si tratamentului
- Markeri pentru sindromul antifosfolipidic
- Serologie boli infectioase
- Teste specializate de alergologie si imunologie
- Teste de biologie moleculara
- Teste de citogenetica
- Teste de microbiologie
- Toxicologie
- Citologie cervico-vaginala
- Histopatologie
- Uncategorized

Beta-talasemie-testare genetica (HBB)
 sânge venos
sânge venos
Beta-talasemiile reprezinta un grup de afectiuni cu transmitere genetica autozomal-recesiva caracterizate printr-o rata scazuta de sinteza a lanturilor β ale globinei.
Clusterul genelor β este localizat pe cromozomul 11, se intinde pe o lungime de 50 kb si contine in urmatoarea ordine: o gena epsilon (ε), 2 gene gamma (denumite Gγ si Aγ), o pseudogena β (ψβ), o gena delta (δ) si o gena β (denumita HBB) care codifica β -globina1. Fiecare gena prezinta 3 secvente care codifica proteine (exoni) si 2 regiuni care aparent nu au nici un rol in sinteza proteinelor (introni = intervening sequences), flancate de secvente 5’ si 3’, care de asemenea nu intervin in procesul de translatie. La capatul 5’ al fiecarei gene se gaseste promotorul, o secventa necesara pentru initierea transcriptiei. Promotorul genei β include 3 secvente: TATA box, CCATT box si motive CACC duplicate. Atat exonii cat si intronii sufera fenomenul de transcriptie a ADN-ului in ARN mesager; acest proces este controlat de factori de transcriptie care se leaga de promotori si de elementele de control situate in amonte – intensificatori (enhancers) – care cresc foarte mult viteza de transcriptie. Astfel, genele β-globinei se afla sub influenta unui element intensificator major, denumit regiunea de control a locusului β (β-LCR) (vezi figura)1;3.
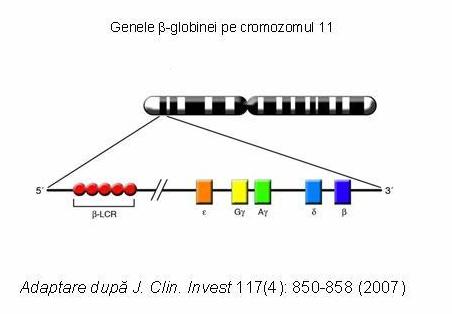

Fenomenul de transcriptie include urmatoarele etape:
– transcriptia propriu-zisa: rezulta o copie ARN a genei, ce include secventele complementare atat pentru exoni cat si pentru introni;
– incapsularea si poliadenilarea (capping + polyadenilation): atasarea unor nucleotide speciale la capetele 5’ si 3’ ale ARN-ului;
– innadirea (splicing): indepartarea intronilor din ARNm; secventele ARN corespunzatoare exonilor sunt apoi “innadite” impreuna3.
Beta talasemia apare mai frecvent la populatiile din bazinul mediteranean insa pot fi afectate si alte grupuri etnice. Severitatea defectului este variabila. Persoanele normale prezinta 2 alele pentru β-globina, astfel ca β-talasemia poate sa apara intr-o stare heterozigota sau homozigota. Deoarece exista un numar mare de mutatii asociate cu β-talasemia sunt posibile si cazuri de heterozigotie compusa, care apar atunci cand o persoana poseda 2 gene mutante.
Sunt descrise doua categorii mari de β-talasemie:
–βo talasemia (beta zero talasemia): caracterizata prin absenta expresiei unei gene β anormale sau mai rar, deletia acesteia; atunci cand defectul apare in stare homozigota (βo/βo) este afectata complet sinteza lanturilor β si implicit a hemoglobinei A;
–β+ talasemie (beta plus talasemie): caracterizata printr-o expresie redusa (dar nu absenta) a unei gene β anormale; atunci cand defectul apare in stare homozigota exista o oarecare productie de hemoglobina A1;5.
Heterozigotii compusi pentru β-talasemie pot avea 2 gene βo sau β+ diferite sau atat o gena β+ cat si una β0 (β+/βo).
Tabloul clinic este extrem de variabil. Statusul heterozigot pentru β-talasemie, denumit si tara β-talasemica sau β-talasemie minora, este in majoritatea cazurilor o conditie clinic asimptomatica. Statusul homozigot sau heterozigotia compusa pentru β-talasemie conduc de obicei la un fenotip clinic sever, denumit β-talasemie majora, in care persoanele afectate sunt dependente de transfuziile de sange. Termenul de β-talasemie intermedia include conditii clinice de severitate intermediara intre formele minora si majora; persoanele afectate sunt simptomatice si pot necesita ocazional transfuzii de sange, fara ca acestea sa fie insa esentiale pentru mentinerea vietii1.
Testarea genetica
Sunt cunoscute la ora actuala peste 200 de mutatii asociate cu β-talasemia, care pot fi detectate prin diverse tehnici PCR, hibridizare cu oligonucleotide sintetice sau secventiere6.
In majoritatea cazurilor este vorba de mutatii aparute la nivelul genei HBB sau in apropierea acesteia (substitutii ale unei singure nucleotide, deletii sau insertii oligonucleotidice). In functie de evenimentul genetic afectat sunt descrise trei tipuri de mutatii:
-care afecteaza transcriptia β-globinei;
-care afecteaza procesarea ARNm (fenomenele de splicing sau poliadenilare);
-care produc o translatie anormala a ARNm (mutatii nonsens sau ale cadrului de citire)3.
Un procent redus de cazuri rezulta ca urmare a unei deletii mari a genei sau a secventelor de control situate in amonte1;2.
Din punct de vedere al severitatii defectului indus mutatiile genei HBB se clasifica in doua mari categorii:
- Mutatii care blocheaza sinteza β-globinei si determina βo-talasemie
- Mutatii nonsens: substitutii ale unei singure nucleotide in codoni normali care creeaza un codon de terminalizare; de exemplu mutatia codonului 39 (C – T) introduce codonul de terminalizare AUG in gena, respectiv rezultand un ARNm nefunctional; aceasta mutatie s-a descoperit la marea majoritate a cromozomilor talasemici de la pacienti din regiunea Mediteraneana.
- Mutatia codonului de initiere: impiedica complet transcriptia; sunt descrise 7 mutatii ale codonului de initiere ATG care codifica metionina.
- Mutatiile cadrului de citire a genei: deletii sau insertii a 1, 2 sau mai mult de 3 nucleotide care altereaza cadrul de citire al ARNm prin introducerea unui codon de terminalizare; de exemplu -1 in codonul 41 (UGA); -1 in codonul 6.
- Mutatiile la nivelul jontiunii de splicing: toate jonctiunile intron – exon confirma regula Chambon (5’ GT/AG 3’) necesara pentru un splicing normal; mutatiile ce altereaza aceste nucleotide nu vor avea un splicing normal, de exemplu IVSI-1 (G – A); IVSII-1 (G – A)6.
- Mutatii care diminueaza sinteza β-globinei si determina β+-talasemie
- Mutatii ale secventei consens: creeaza situsuri de splicing alternative, de exemplu: IVSI-5 (G – T); IVSI-6 (T – C); ultima mutatie este de origine mediteraneana.
- Mutatii punctiforme in exoni: mutatiile in secventele codificatoare pot determina procesari anormale ale ARNm; secventa din jurul dinucleotidului GT din codonul 25 are 6 din 9 pozitii in comun cu secventa consens de la situsul de jonctiune de la capatul 5’, dar nu par sa fie implicate in procesarea normala; au fost descrise trei mutatii in interiorul acestui situs de splicing care duc la activarea sa: prima mutatie (G®A din codonul 26) da nastere structurii proprii HbE care este asociata cu fenotipul unei β-talasemii usoare; similar, Hb Knossos are o mutatie in codonul 27 (G®T) si prezinta fenotip β-talasemic; a treia mutatie ce face parte din acest tip este substitutia A®T din codonul 24 – o substitutie silentioasa la nivel de proteina, dar asociata cu o severitate moderata a fenotipului β+-talasemic.
- Mutatii punctiforme in introni: produc situsuri criptice de splicing si astfel se genereaza un ARNm aberant in plus fata de un ARNm normal; de exemplu IVSI-110, IVSII-745.
- Mutatii ale promotorului: au fost descrise diverse mutatii in regiunea promotorului care reduc rata de legare a ARN polimerazei, ceea ce are ca rezultat reducerea transcriptiei cu 20-30%; mutatiile 28 A→ G si 29 A→G sunt frecvente la populatia chineza si africana, in timp ce mutatiile – 87 C→G si silent – 101 C→T sunt mai frecvente la populatia din bazinul mediteranean1;6.
Recomandari pentru analiza mutatiilor
-stabilirea prognosticului in ceea ce priveste fenotipul clinic (diferentierea moleculara a talasemiei intermedia de forma majora);
-depistarea purtatorilor in familiile cu risc crescut;
-diagnosticul prenatal al beta talasemiei5;6.
Specimen recoltat – sange venos4.
Recipient de recoltare – vacutainer ce contine EDTA ca anticoagulant4.
Cantitate recoltata – cat permite vacuumul4
Cauze de respingere a probei – folosirea heparinei ca anticoagulant; probe coagulate sau hemolizate4.
Stabilitate proba – 7 zile la 2-8ºC4.
Metoda – secventiere completa HBB4.
Interpretarea rezultatelor
Tara de beta-talasemie (statusul heterozigot)
Persoanele purtatoare ale tarei de β-talasemie sunt de obicei asimptomatice; este posibil insa ca in conditii de stres hematopoietic (sarcina, infectii intercurente) sa apara manifestari legate de anemie. Unii pacienti pot sa aiba splenomegalie.
Hemograma prezinta modificari caracteristice ale seriei eritrocitare: scadere usoara a valorii hemoglobinei, numar crescut de eritrocite, valori scazute ale indicilor eritrocitari VEM si HEM, largime a distributiei eritrocitare de obicei normala. In medie, variabilele hematologice difera la tarele β0 si β+, desi poate exista o suprapunere considerabila. In cazurile in care coexista α-talasemia anomaliile hematologice sunt mai putin marcate.
Pe frotiul de sange periferic, alaturi de microcitoza, se pot observa hipocromie, poikilocitoza, hematii “in tinta”, punctatii bazofile. La examenul maduvei osoase se constata o celularitate crescuta ca urmare a hiperplaziei eritroide.
Diagnosticul se bazeaza pe detectarea unui procent crescut de HbA2 la electroforeza hemoglobinei sau prin tehnica cromatografica (HPLC). Aproximativ o treime-o jumatate din pacienti asociaza si un procent crescut de HbF.
Proportia de hemoglobina A2 se coreleaza cu tipul mutatiei beta-talasemice. Astfel, in majoritatea cazurilor de heterozigotie pentru β0 sau de β+-talasemie severa se inregistreaza un procent de 4-5% HbA2, in timp ce in formele usoare de β+-talasemie procentul este de obicei de 3.6-4.2%. Procente mai mari se constata in cazul tarelor asociate cu deletia portiunii 5’ a genei HBB, precum si in cazurile de mutatii ale promotorului sau ale codonului de initiere.
Cauze de tara β-talasemica cu HbA2 normala
Exista si forme de β-talasemie cu un procent normal de HbA2, denumite silent β-thalasemmia. De exemplu in cazul mutatiei C→G din pozitia 6, 3’ la codonul de terminalizare se constata un procent mediu de 2.4%.
Daca o persoana prezinta indici eritrocitari sugestivi pentru tara beta-talasemica dar procentul de HbA2 nu este crescut este important sa se excluda δβ-talasemia, hemoglobinopatia Lepore, precum si α-talasemia. Poate sa existe si o γδβ-talasemie, insa aceasta conditie este foarte rara. Unii pacienti cu anemie feripriva pot sa aiba indici eritrocitari atipici; in special copiii pot prezenta o anemie feripriva insotita de un numar crescut de eritrocite.
Un nivel scazut de feritina indica un deficit de fier dar nu exclude diagnosticul de tara beta-talasemica.
Mutatiile HBB asociate cu anomalii hematologice minore sau absente in statusul heterozigot pun probleme importante de diagnostic; identificarea acestora este necesara deoarece pot determina o afectiune semnificativa clinic la homozigoti sau heterozigoti compusi. Aceste tipuri de mutatii pot fi impartite in doua grupuri:
-mutatii care determina “silent thalassemia” cu indici eritrocitari si procent de HbA2 normale;
-mutatii care determina “almost silent thalassemia” cu procent de HbA2 normal si indici eritrocitari modificati.
Aceste forme pot fi detectate numai prin testare genetica. Unele mutatii, cum ar fi +1480 C→G intalnita la populatia greaca, sunt atat de usoare incat determina aproape intotdeauna “silent thalassemia”. Mutatiile cel mai frecvent responsabile de aceasta forma de talasemie sunt -101 C→T si – 92 C→T asociate cu urmatoarele valori medii: VEM=83.9 fL, HEM=28.6 pg si HbA2=3.4%. “Almost silent thalassemia” rezulta dintr-un grup mici de mutatii, cum ar fi CAP+1 A→C in India si ocazional IVS-6 T→C la mediteraneeni. Heterozigotii pentru IVS-6 T→C prezinta urmatoarele intervale de valori : VEM=60-78 fL si HbA2=3.2-4-6%.
Deficitul de fier reduce procentul de HbA2 atat la persoanele fara defecte ale globinei cat si la cele cu tara beta-talasemica. Acest lucru nu reprezinta o problema pentru persoanele cu o anomalie genetica insotita de cresteri semnificative de HbA2, insa pentru cei cu mutatii usoare asociate cu cresteri minore de HbA2 deficitul de fier mascheaza tara beta-talasemica. Din acest motiv se recomanda ca la pacientii cu anemie feripriva severa sau moderata sa se corecteze mai intai deficitul de fier dupa care, in cazul in care persista modificarile indicilor eritrocitari, sa se masoare procentul de HbA2. O exceptie de la aceasta recomandare se va face in cazul pacientelor gravide la care diagnosticul tarei beta-talasemice trebuie stabilit imediat. In aceasta situatie este necesar sa se investigheze si partenerul; daca acesta este depistat cu tara beta-talasemica, se va lua in considerare testarea genetica la gravidele cu niveluri de HbA2 la limita.
Anemia megaloblastica poate conduce de asemenea la reducerea procentului de HbA2 si interfera cu diagnosticul tarei β-talasemice.
Asocierea indicilor eritrocitari modificati cu un nivel normal de HbA2 poate rezulta si din coexistenta δ-talasemiei cu o mutatie β0 sau β+ ce induce un procent mare de HbA2 (intalnita la sarzi si ciprioti). Hemoglobina Knossos este responsabila de asemenea de unele cazuri de tara β-talasemica cu un nivel normal de HbA2.
Prezenta concomitenta a unei mutatii β-talasemice usoare (cum ar fi CAP+1 A→C) si a unei tare de α0-talasemie sau a unui status homozigot pentru α+-talasemie face ca diagnosticul tarei β-talasemice sa fie adesea omis.
Tara β-talasemica cu indici eritrocitari normali si HbA2 crescuta
Unii pacienti heterozigoti pentru anumite variante de β-talasemie usoara pot sa aiba niveluri crescute de HbA2 in prezenta unor indici eritrocitari normali; aceasta conditie a fost constata la o parte din pacientii purtatori ai mutatiei -101 C→T.
In plus, coexistenta α si β-talasemiei poate conduce la indici eritrocitari normali printr-o sinteza mai echilibrata a lanturilor globinei, insa cu un procent crescut de HbA2. Aceasta modificare este intalnita in special la pacientii avand deletii a doua gene α sau o forma de α-talasemie nedeletionala.
Anomaliile dobandite, cum ar fi cele induse de afectiuni hepatice, pot creste valorile indicilor eritrocitari la pacientii cu tara β-talasemica.
Toate aceste exemple ilustreaza complexitatea cazurilor de tara β-talasemica si dificultatea identificarii acestora, testarea genetica fiind uneori singura investigatie care poate confirma diagnosticului1;2.
Beta-talasemia dominanta
Desi majoritatea persoanelor heterozigote pentru o mutatie β-talasemica prezinta caracteristici clinicopatologice descrise ca talasemie minora (tablou hematologic modificat in absenta manifestarilor clinice), unele dintre acestea dezvolta splenomegalie, anemie, icter si litiaza biliara, conditie denumita β-talasemie dominanta. Fenotipul este similar β-talasemiei intermedia si este asociat atat cu o eritropoieza ineficienta cat si cu o componenta hemolitica. Prezenta eritroblastilor cu incluzii (compuse din lanturi α in exces si lanturi β-anormale) este caracteristica.
Sunt descrise peste 30 alele care induc fenotipul de β-talasemie dominanta, printre care se numara mutatiile aparute la nivelul regiunii 3’ a exonilor 2 sau 3 care determina o cantitate importanta de ARNm mutant. Ca o consecinta, se sintetizeaza lanturi β trunchiate sau alungite, extrem de instabile, care co-precipita cu lanturile α normale1.
Beta-talasemia intermedia
Se refera la un fenotip clinic determinat de anomalii genetice diverse. Severitatea β-talasemiei intermedia variaza de la o conditie clinica insotita de retard de crestere si deformatii osoase, in care supravietuirea este rareori posibila fara transfuzii de sange, pana la forme mai usoare, asemanatoare tarei β-talasemice, dar asociate cu un grad mai mare de anemie si splenomegalie.
Pot sa apara complicatii cum ar fi: hipersplenism, litiaza biliara, supraincarcare cu fier, fenomene de compresiune exercitate de masele de tesut hematopoietic extramedular asupra unor organe vitale.
Tabloul hematologic include anomalii similare celor induse de β-talasemia heterozigota tipica, insa are o severitate mai mare; alaturi de microcitoza, hipocromie, anizocitoza, poikilocitoza, punctatii bazofilie, pot sa apara policromatofilie si eritroblasti circulanti.
Electroforeza hemoglobinei sau HPLC indica niveluri crescute de HbA2 (mai mari decat cele intalnite in tara β-talasemica) si HbF.
β-talasemia intermedia poate sa apara la pacientii care prezinta fie una, fie ambele gene β anormale. Cei care sunt homozigoti sau heterozigoti compusi pentru alele β-talasemice au mutatii asociate cu forme usoare de β+-talasemie sau prezinta factori amelioratori, cum ar fi coexistenta tarei α-talasemice sau o mutatie care conduce la o sinteza crescuta de lanturi γ in conditii de stres hematopoietic. Acele persoane care au doar o gena β anormala fie prezinta o forma de β-talasemie dominanta, fie asociaza concomitent mutatii care agraveaza dezechilibrul lanturilor globinei, cum ar fi statusul homozigot sau heterozigot pentru triplu α sau cvadruplu α (gene α multiplicate de 3 sau 4 ori).
Testele genetice faciliteaza identificarea anomaliilor care determina fenotipul de β-talasemie intermedia1;5.
Beta-talasemia majora
Pacientii homozigoti sau heterozigoti compusi pentru mutatii β-talasemice care sunt dependenti de transfuzii de sange prezinta conditia clinica numita β-talasemie majora.
Boala este recunoscuta adesea in primul an de viata, incepand cu varsta de 3 luni; se caracterizeaza printr-o anemie severa insotita de o eritropoieza accelerata, atat in sectorul medular cat si in cel extramedular. Expansiunea hematopoietica conduce la deformari osoase severe, in special ale oaselor craniului si fetei; hematopoieza extramedulara conduce la hepatosplenomegalie masiva.
Seria eritrocitara prezinta urmatoarele valori: Hb=3-7 g/dL, VEM=50-60 fL, HEM=12-18 pg. Frotiul de sange include microcitoza, hipocromie, anizocitoza si poikilocitoza marcate, punctatii bazofile, eritroblasti circulanti cu defect de hemoglobinizare si trasaturi diseritropoietice.
In cazurile de homozigotie sau heterozigotie compusa pentru β0-talasemia (β0/β0) electroforeza hemoglobinei sau tehnica HPLC indica prezenta numai de HbA2 si HbF; atunci cand pacientii prezinta homozigotie pentru β+-talasemie (β+/β+) sau heterozigotie compusa pentru β0 si β+-talasemie (β+/β0) este prezenta si HbA intr-un procent de pana la 35%.
In β-talasemia majora procentul de HbA2 poate fi normal, crescut sau, ocazional, scazut1;2.
Bibliografie
1. Barbara J. Bain. Haemoglobinopathy Diagnosis. Blackwell Science, 2001, 7-9; 73-93.
2. Caterina Borgna Pignatti, Renzo Galanello. Thalassemias and Related Disorders of Hemoglobin Synthesis. In Wintrobe`s Clinical Hematology, 11th ed, Lippincott Williams & Wilkins, 2004, 1083-1118.
3. Cornel Mironel Niculae. Transcrierea (transcriptia). Bioinformatica. fpce9.fizica.unibuc.ro/bioinfo/transcrierea.htm. Reference Type: Internet Communication.
4. Laborator Synevo. Referintele specifice tehnologiei de lucru utilizate 2010. Ref Type: Catalog.
5. Renzo Galanello, Antonio Cao. Beta-Thalassemia. Gene Reviews, 2007. www.ncbi.nlm.nih.gov. ReferenceType: Internet Communication.
6. Talmaci R, Diagnosticul molecular al -talasemiilor. Progrese in biotehnologie, vol. II. Ed. Ars Docendi, p.179-188, 2002.